重症筋無力症
(MG:Myasthenia Gravis)
免疫系の異常により、自分の細胞を間違って攻撃してしまう自己免疫疾患のひとつです。
末梢神経系と筋肉の接合部が破壊され、筋肉への刺激伝達が難しくなる病気です。発症に関わるアセチルコリン受容体(AChR)抗体と、筋特異的受容体型チロシンキナーゼ(MuSK)抗体という自己抗体が発見されています。
疫学(日本)
- 患者数 約29,210人1)
- 有病率 23.1人/10万人1)
- 発症年齢(中央値) 59歳2)
- 男女比 1:1.152)
原因(病態)
免疫系の異常により、末梢神経系と筋肉の接合部において、筋肉側が自己抗体の攻撃を受けて破壊され、筋肉への刺激がスムーズに伝わらなくなる病気です(図1)。
約80~85%のMG患者さんがAChR抗体陽性、約5%がMuSK抗体陽性の患者さんです。残りの約10~15%は両抗体(AChR抗体・MuSK抗体)陰性の患者さんです1)。
図1:MGの原因(イメージ図)
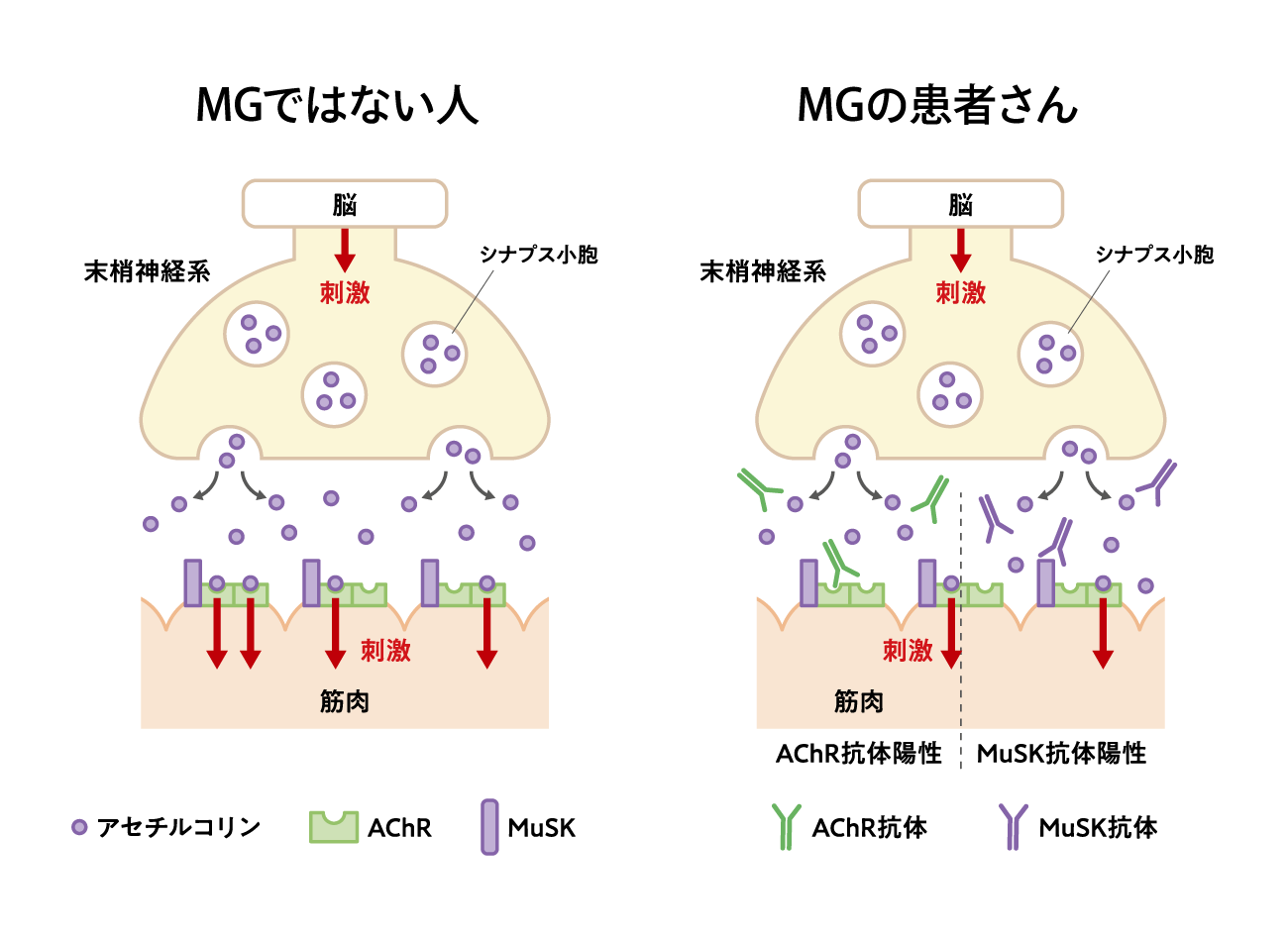
末梢神経系:中枢神経系(脳・脊髄)以外の神経系で、脳や脊髄から伸び出る神経のこと(脳神経、脊髄神経とも呼ばれます)。中枢神経系から発信された指令を体の各部に伝える働きを担っています。
シナプス小胞:神経細胞のシナプス末端で、神経伝達物質を貯蔵している小胞。
アセチルコリン受容体(AChR):神経と筋肉の接合部で、神経伝達物質アセチルコリンに応答する、筋肉側に存在する受容体。
筋特異的受容体型チロシンキナーゼ(MuSK):神経と筋肉の接合部で、筋肉側に存在する膜貫通型のタンパク質。
症状
MGは、運動などにより体の筋肉が疲れやすく、休憩することで改善します。また、夕方に悪化したり(日内変動)、日によって症状が変わったりする(日差変動)などの特徴があります。 どの筋肉が侵されるかによって、あらわれる症状はさまざまで(図2)、原因となった自己抗体の種類によって筋力低下の程度や部位も異なります1)。
図2:MGの症状の特徴1)
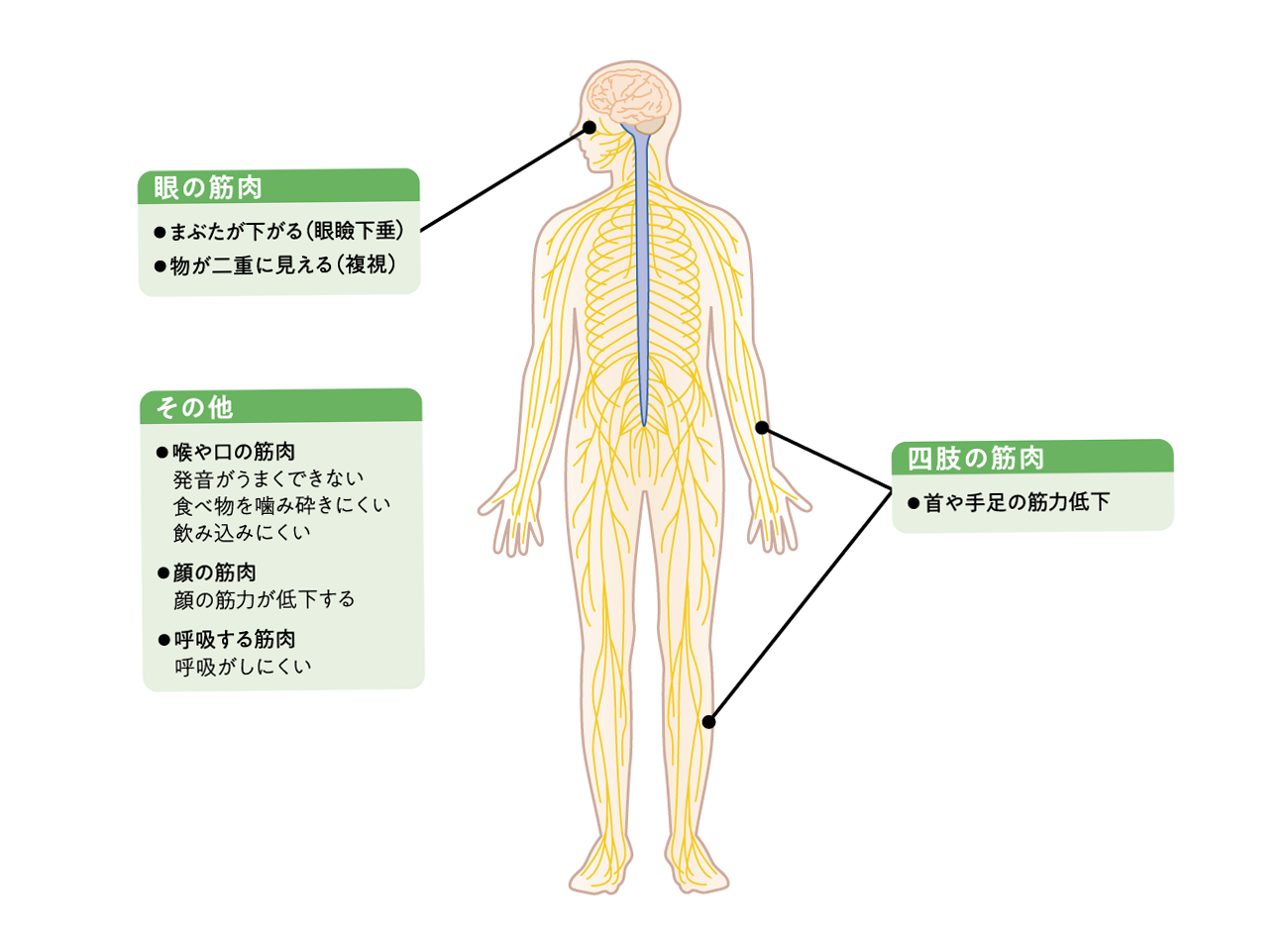
なかには、胸腺の異常(胸腺腫など)が合併する患者さんや、他の自己免疫疾患であるバセドウ病、慢性甲状腺炎(橋本病)、関節リウマチなどの合併症を抱えている患者さんもいます。
見た目ではわからない症状も多く、人から理解してもらえないことが多いため、孤独を感じる患者さんも少なくありません。
経過
治療の進歩によって、重症化する患者さんは激減しています。
成人発症のMG患者さんは、症状がなくなった状態(寛解)を長期にわたって完全に維持することはまだ難しいものの、適切な治療を継続することで、半数以上の患者さんが、生活や仕事に支障がない状態まで改善しています。完全に治療が不要になる方は6%程度で、10~20%の方は治療による改善があまりない難治性です2)。
MG患者さんは、臨床症状、病原性の自己抗体や胸腺異常の有無などによって、眼筋型と5つの全身型、6タイプに分類されます(図3)1)。
図3:MGの病型1)

眼筋型から全身型へ移行する患者さんは20%で、多くは発症2年以内に移行します1, 3)。眼筋型は、全身型に比べて軽症とされますが、患者さんは日常生活において多くの悩みを抱えています。
【出典】
1)重症筋無力症/ランバート・イートン筋無力症候群診療ガイドライン2022. p. 2-4, 14-17, 31-33,
36-43, 50-53.
2)難病情報センターホームページ(2024年4月現在)
3)Akaishi T et al. PLoS One. 2014; 9(9): e106757.